



CRISPR Knockin Stable Cell Line
VectorBuilder can engineer stable cell lines with permanent knockin of your gene of interest (GOI) at desired genomic target sites. Stable knockin is achieved via a CRISPR-based approach wherein a target site specific gRNA/Cas9 RNP complex is introduced into the target cells along with a donor vector carrying the template for homology-directed repair (HDR). Our proprietary technology allows us to achieve very efficient gene delivery and HDR, resulting in rapid turnaround and high successful rate for obtaining homozygous knockin.
Highlights
- Supreme integration efficiency: most cells exhibit HDR rate of 30-50% with our proprietary donor design and gene delivery approach.
- Large fragment knockin: fragments of up to 1 kb can be efficiently introduced into a variety of cells, including ESCs and iPSCs.
- Rapid turnaround: monoclonal knockin cells can be delivered in as fast as 16 weeks starting from vector design.
Service Details

Figure 1. Typical workflow of generating gene knockin cells by CRISPR.
Price and turnaround
| Service Type | Deliverable | Price (USD)* | Turnaround |
|---|---|---|---|
| Knockin (fragment<3 kb) | One heterozygous single clone (>106 cells/vial, 2 vials) | From $7,999 | 14-20 weeks |
| One homozygous single clone (>106 cells/vial, 2 vials) | Please inquire | ||
| Knockin (fragment>3 kb) | One heterozygous single clone (>106 cells/vial, 2 vials) | From $10,999 | 16-24 weeks |
| One homozygous single clone (>106 cells/vial, 2 vials) | Please inquire | ||
* Additional charge will apply for extra single clones or vials.
QC assays
| Assay | Methods |
|---|---|
| Knockin validation (default) | Genotyping PCR, Sanger sequencing |
| Expression test (add-on) | RT-qPCR, WB, IF, FACS |
| Off-target analysis (add-on) | NGS, PCR, Sanger sequencing |
| Chromosome analysis (add-on) | Karyotyping |
| Sterility (default) | PCR for mycoplasma detection, bioburden test for sterility detection |
Downstream services
We can perform various phenotypic assessments and functional validation of your engineered cell lines, including assays for proliferation, apoptosis, migration, viability, cytotoxicity, etc.
Case Studies

Figure 2. CRISPR-mediated gene knockin in iPSCs. Knockin of UBC-driven EGFP (2432 bp) into iPSCs was achieved by electroporation of Cas9/gRNA RNP complex and donor vector. (A) Confirmation of EGFP knockin at target site by Sanger sequencing. (B) Genotyping PCR of four single clones with homozygous knockin. The WT locus is 762 bp and the locus with EGFP knockin is 3194 bp. (C) EGFP fluorescence in knockin cells by microscopy. (D) Karyotyping results. (E) Expression of pluripotency markers NANOG, OCT4, and SOX2 in EGFP knockin iPSCs by immunofluorescence.
How to Order

Resources
FAQ
Gene knockout relies on non-homologous end joining (NHEJ) mediated DNA repair of double strand breaks (DSBs) caused by CRISPR which is error-prone. As a result, mutations such as frameshift or fragment deletion (when using dual-gRNA) happen at a certain frequency which cause loss-of-function of the target gene. Differently, gene knockin depends on precise insertion of donor fragment at target site by HDR which is much less efficient than NHEJ and is limited to dividing cells. Additionally, HDR efficiencies vary by cell type. Altogether, gene knockin is technically more challenging than gene knockout.
The primary consideration for the format of your template is size. If introducing a small insert (<10 bp), then ssODNs are typically the best option because of its enhanced efficiency of HDR. Larger templates must be introduced with dsDNA in either circular plasmid form (higher cell tolerance but lower HDR efficiency) or linear form (higher HDR efficiency but higher cell toxicity).
Following transfection and antibiotic drug selection, a pool of positively transfected cells is established. Cells are counted, e.g. with a hemocytometer, to determine cell number and concentration. Cells then go through serial dilution (limiting dilution) and are cultured in 96-well plates at a concentration of <1 cell per well. These individual viable clones are expanded and genotyped to confirm the desired genetic modification.
Featured Citations
The KU-PARP14 axis differentially regulates DNA resection at stalled replication forks by MRE11 and EXO1
Ashna Dhoonmoon, Claudia M. Nicolae & George-Lucian Moldovan.
Nature Communications, 2022, doi: 10.1038/s41467-022-32756-5
Products used:
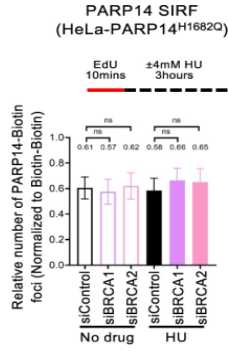
Abstract: Suppression of nascent DNA degradation has emerged as an essential role of the BRCA pathway in genome protection.In BRCA-deficient cells, the MRE11 nuclease is responsible for both resection of reversed replication forks, and accumulation of single stranded DNA gaps behind forks. Here, we show that the mono-ADP-ribosyltransferase PARP14 is a critical co-factor of MRE11. PARP14 is recruited to nascent DNA upon replication stress in BRCA-deficient cells, and through its catalytic activity, mediates the engagement of MRE11. Loss or inhibition of PARP14 suppresses MRE11-mediated fork degradation and gap accumulation, and promotes genome stability and chemoresistance of BRCA-deficient cells. Moreover, we show that the KU complex binds reversed forks and protects them against EXO1-catalyzed degradation. KU recruits the PARP14-MRE11 complex, which initiates partial resection to release KU and allow long-range resection by EXO1. Our work identifies a multistep process of nascent DNA processing at stalled replication forks in BRCA-deficient cells.
>See more<Collapse 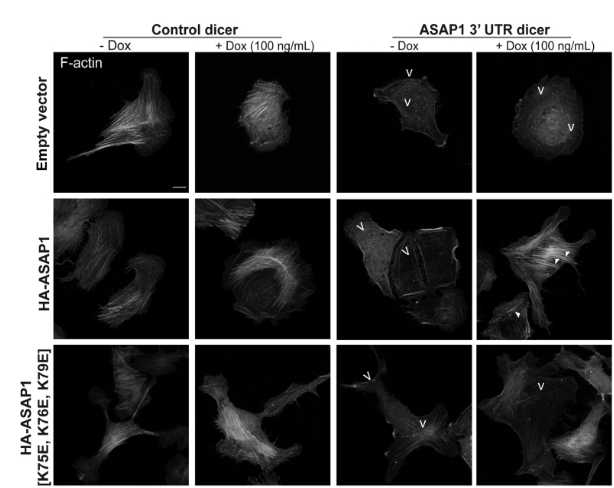
A lysine-rich cluster in the N-BAR domain of ARF GTPase-activating protein ASAP1 is necessary for binding and bundling actin filaments
Anjelika Gasilina, et al.
Journal of Biological Chemistry, 2022, doi: 10.1016/j.jbc.2022.101700
Products used:
Abstract: Actin filament maintenance is critical for both normal cell homeostasis and events associated with malignant transformation. The ADP-ribosylation factor GTPase-activating protein ASAP1 regulates the dynamics of filamentous actin_x0002_based structures, including stress fibers, focal adhesions, and circular dorsal ruffles. Here, we have examined the molecular basis for ASAP1 association with actin. Using a combination of structural modeling, mutagenesis, and in vitro and cell-based assays, we identify a putative-binding interface between the N-Bin-Amphiphysin-Rvs (BAR) domain of ASAP1 and actin filaments. We found that neutralization of charges and charge reversal at positions 75, 76, and 79 of ASAP1 reduced the binding of ASAP1 BAR-pleckstrin homology tandem to actin filaments and abrogated actin bundle formation in vitro. In addition, overexpression of actin-binding defective ASAP1 BAR-pleckstrin homology [K75, K76, K79] mutants prevented cellular actin remodeling in U2OS cells. Exogenous expression of [K75E, K76E, K79E] mutant of full-length ASAP1 did not rescue the reduction of cellular actin fibers consequent to knockdown of endogenous ASAP1. Taken together, our results support the hypothesis that the lysine-rich cluster in the N-BAR domain of ASAP1 is important for regulating actin filament organization.
>See more<Collapse