



CRISPR Genome Editing Solutions
VectorBuilder offers a comprehensive collection of CRISPR products and services to provide you with the ideal tools for in vitro and in vivo genome editing experiments. Our CRISPR offerings range from off-the-shelf reagents that are ready for transfection or transduction to custom-made CRISPR vectors with high targeting efficiency and available in multiple delivery formats (i.e. plasmid, virus, RNA). We can package all major virus types (i.e. lentivirus, AAV, adenovirus) to deliver your CRISPR components efficiently into difficult-to-transfect cells. Additionally, we are specialized in building high-quality CRISPR libraries for knockout, gene activation, gene inhibition and other CRISPR screens. Our uniquely designed and well validated whole genome dual-gRNA knockout libraries are powerful tools for gene functional screens in human and mouse.
Click to view user testimonials about our CRISPR genome editing services
Highlights
- Highly intuitive online vector design platform with whole-genome gRNA database implemented for easy and quick CRISPR design
- Rich collection of vector backbones and vector components
- CRISPR components available in a variety of delivery formats (i.e. CRISPR/Cas9 plasmid, CRISPR/Cas9 virus, Cas9 mRNA + gRNA, Cas9-gRNA RNP complex)
- Versatile CRISPR library construction options
- Robust quality, fast turnaround, and competitive pricing
- Powerful technical support for experimental design, data analysis, and troubleshooting
Offering Details
- Custom CRISPR vectors
- Popular CRISPR vectors
- CRISPR virus
- RNA preparation for Cas9 mRNA and gRNA
- Donor DNA for precise genome editing
- Pooled CRISPR libraries
- CRISPR solutions
Technical Information
- CRISPR-mediated genome editing
- CRISPR-mediated gene regulation
- CRISPR delivery approaches
- gRNA databases
Offering Details Price Match
Custom CRISPR vectors
CRISPR-mediated genome editing requires target cells to co-express Cas9 and target site-specific gRNA at the same time. Using our highly intuitive online vector design platform, you can choose from over 30 vector backbones (non-viral, viral or transposon) and unlimited combinations of vector components (promoters, Cas9 variants, fluorescent and drug-selection markers) to express Cas9 and your gRNA(s) using different approaches. Furthermore, we offer all-in-one as well as dual vector systems for Cas9 and gRNA expression in regular plasmid, lentivirus, AAV, adenovirus and PiggyBac backbones.
Our comprehensive collection of CRISPR vectors also includes gene targeting donor vectors serving as DNA templates to guide precise sequence changes such as point mutations and large fragment knockins at target sites via HDR. Additionally, we offer CRISPRa and CRISPRi vectors for achieving transcriptional activation or inhibition of target genes, respectively, within their endogenous loci. We can design and construct vectors for dCas9-SAM-based or dCas9-SunTag-based CRISPR activation and dCas9-KRAB or dCas9-KRAB-MeCP2 based CRISPR inhibition applications.
Plasmid DNA preparation, RNA preparation and virus packaging can be purchased as downstream services when adding CRISPR vectors into your shopping cart.
Design tips
Note: AAV has a cargo capacity of 4.7 kb. The commonly used SpCas9 derived from Streptococcus pyogenes is 4.2 kb, which can barely fit into AAVs when combined with other components such as a promoter, the polyA signal sequence, and the gRNA expression cassette. Our standard AAV CRISPR system therefore utilizes the shorter SaCas9 (3.2 kb) derived from Staphylococcus aureus. Please note that the PAM sequence recognized by SaCas9 is NNGRR or NNGRRT (preferred), whereas the PAM for SpCas9 is NGG. We can also construct AAV SpCas9 vectors upon request.
Choose your custom vector View more
Popular CRISPR vectors
In addition to custom CRISPR/Cas9 vectors, VectorBuilder also offers a panel of premade Cas9 vectors, control gRNA vectors and helper vectors for CRISPRa and CRISPRi. Our off-the-shelf vectors are available for immediate shipment as E. coli stocks. Plasmid DNA preparation, RNA preparation and virus packaging can be purchased as downstream services upon adding premade vectors into your shopping cart.
Click here to view our collection of premade CRISPR vectorsView more
| Vector System | Vector Name | Vector ID |
|---|---|---|
| hCas9 expression vector (regular plasmid) | pRP[Exp]- mCherry/Hygro-CBh>hCas9 |
VB010000-9378bvk |
| hCas9 expression vector (lentivirus) | pLV[Exp]- CBh>hCas9/Hygro |
VB010000-9380sne |
| SaCas9 expression vector (AAV) | pAAV[Exp]-CMV>SaCas9 | VB010000-9382per |
| hCas9 expression vector (adenovirus) | pAV[Exp]-CBh>hCas9 | VB010000-9381pwj |
| hCas9 expression vector (piggyBac) | pPB[Exp]-mCherry/Hygro-CBh>hCas9 | VB010000-9379gqq |
| Scramble gRNA control vector (regular plasmid) | pRP[gRNA]-EGFP/Puro-U6>Scramble_gRNA | VB010000-9358ttk |
| Scramble gRNA control vector (lentivirus) | pLV[gRNA]-EGFP/Puro-U6>Scramble_gRNA | VB010000-9359hhe |
| Scramble SagRNA control vector (AAV) | pAAV[SagRNA]-EGFP-U6>Scramble_SagRNA1 | VB010000-9361zjr |
| Scramble gRNA control vector (adenovirus) | pAV[gRNA]-EGFP-U6>Scramble_gRNA1 | VB010000-9360nph |
| Scramble gRNA control vector (piggyBac) | pPB[gRNA]-EGFP/Puro-U6>Scramble_gRNA1 | VB010000-9362zuk |
| hCas9 and scramble gRNA coexpression vector (regular plasmid) | pRP[CRISPR]-EGFP/Puro-hCas9-U6>Scramble_gRNA1 | VB010000-9354ztt |
| hCas9 and scramble gRNA coexpression vector (lentivirus) | pLV[CRISPR]-hCas9/Puro-U6>Scramble_gRNA1 | VB010000-9355sqw |
| SaCas9 and scramble SagRNA coexpression vector (AAV) | pAAV[CRISPR]-SaCas9-U6>Scramble_SagRNA | VB010000-9357zmm |
| hCas9 and scramble gRNA coexpression vector (adenovirus) | pAV[CRISPR]-hCas9/EGFP-U6>Scramble_gRNA1 | VB010000-9356pna |
| dCas9-SAM activator MS2/P65/HSF1 expression vector (lentivirus) | pLV[Exp]-EF1A>MS2/P65/HSF1/Hygro | VB010000-9383ffr |
| dCas9-SAM activator dCas9/VP64 expression vector (lentivirus) | pLV[Exp]-EF1A>dCas9/VP64/Bsd | VB010000-9384wwc |
| dCas9-SAM scramble msgRNA control vector (lentivirus) | pLV[msgRNA]-EGFP/Puro-U6>Scramble_gRNA | VB010000-9363gsm |
| dCas9-KRAB-MeCP2 expression vector (lentivirus) | pLV[Exp]-CBh>dCas9/ KRAB/MeCP2/Hygro |
VB010000-9386mwf |
CRISPR virus
Lentivirus, AAV and adenovirus are widely used to deliver CRISPR components into mammalian cells. VectorBuilder offers premium-quality virus packaging services for lentivirus, AAV and adenovirus for achieving highly efficient CRISPR targeting in difficult-to-transfect cells. Our proprietary technologies and reagents have greatly improved virus packaging protocols in terms of titer, purity, viability and consistency. Our packaging protocols are also optimized for the viral vector systems used in our vector construction services. As a result, we have a growing base of highly satisfied customers who come back to us again and again for their cloning and virus packaging needs.
Price, turnaround and scales of lentivirus packaging servicesView more
| Scale | Application | Typical Titer | Minimum Titer | Volume | Price (USD) | Turnaround |
|---|---|---|---|---|---|---|
| Mini | Cell culture | >2x108 TU/ml | >108 TU/ml | 100 ul (4x25 ul) | $199 | 6-12 days |
| Pilot | >4x108 TU/ml | 250 ul (10x25 ul) | $449 | |||
| Medium | >3x108 TU/ml | 1 ml (10x100 ul) | $649 | |||
| Large | >2x109 TU/ml | >109 TU/ml | 1 ml (10x100 ul) | $1,099 | ||
| Ultra-purified medium | Cell culture & in vivo | >2x109 TU/ml | >109 TU/ml | 500 ul (10x50 ul) | $1,399 | |
| Ultra-purified large | 1 ml (10x 100 ul) | $1,699 |
TU = Transduction units (also known as infectious units)
Price, turnaround and scales of AAV packaging services View more
| Scale | Application | Typical Titer | Minimum Titer | Volume | Price (USD) | Turnaround |
|---|---|---|---|---|---|---|
| Pilot | Cell culture | >1012 GC/ml | >2x1011 GC/ml | 250 ul (10x25 ul) | $449 | 6-12 days |
| Medium | 1 ml (10x100 ul) | $649 | ||||
| Large | >5x1012 GC/ml | >2x1012 GC/ml | 1 ml (10x100 ul) | $1,099 | ||
| Ultra-purified pilot | Cell culture & in vivo | >2x1013 GC/ml | >1013 GC/ml | 100 ul (4x25 ul) | $1,399 | 7-14 days |
| Ultra-purified medium | 500 ul (10x50 ul) | $1,999 | ||||
| Ultra-purified large | 1 ml (10x100 ul) | $3,099 |
GC = Genome copies
Price, turnaround and scales of adenovirus packaging servicesView more
| Scale | Application | Typical Titer | Minimum Titer | Volume | Price (USD) | Turnaround |
|---|---|---|---|---|---|---|
| Pilot | Cell culture | >2x1010 IFU/ml | >1010 IFU/ml | 250 ul (10x25 ul) | $649 | 28-35 days |
| Medium | 1 ml (10x100 ul) | $1,099 | ||||
| Large | >2x1011 IFU/ml | >1011 IFU/ml | 1 ml (10x100 ul) | $1,699 | ||
| Ultra-purified medium | Cell culture & in vivo | >2x1012 VP/ml | >1012 VP/ml | 500 ul (10x50 ul) | $2,099 | |
| Ultra-purified large | 1 ml (10x100 ul) | $2,499 |
IFU = Infectious units; VP = Virus particles
Click here to view detailed information on our virus packaging services
RNA preparation for Cas9 mRNA and gRNA
VectorBuilder can provide transfection-ready and micro injection-ready Cas9 mRNA and gRNA specifically designed against user-selected target sites for easy RNA-based delivery of CRISPR components into mammalian cells. Cas9 mRNA is available for both wildtype hCas9 nuclease and Cas9 nickase (Cas9(D10A)). We follow the algorithm utilized in CRISPR library design (CLD) to calculate specificity score and apply a set of empirical rules to design the optimal gRNA for targeting user-selected genes/sites.
Price and turnaround of CRISPR RNA products View more
| Reagent | Concentration & Volume | Price (USD) | Turnaround |
|---|---|---|---|
| hCas9 mRNA | >500 ng/ul, 25 ul, nuclease-free water, sterile | $449 | 2-4 days |
| Cas9(D10A) mRNA | |||
| Custom gRNA* | $349 |
*Cloning of gRNA into in vitro transcription vector has an additional $149 charge and 5-10 days turnaround
Donor DNA for precise genome editing
VectorBuilder offers donor DNA templates in the form of ssODN or dsDNA from linearized plasmid for guiding HDR-based DNA repair to introduce precise DNA sequence changes at CRISPR cleavage sites. The introduced changes include point mutations and small or large fragment knockins. While ssODNs are utilized for inserting short DNA sequences (< 60 bp) such as small tags or restriction enzyme sites at target sites, dsDNA donors are utilized for targeted knockin of larger sequences (up to 4-5 kb) such as fluorescent tags and other reporters.
Price and turnaround of donor DNA productsView more
| Reagent | Price (USD) | Turnaround |
|---|---|---|
| ssODN (normally 120-200 nt) | From $349 | 2-3 weeks |
Pooled CRISPR libraries
VectorBuilder specializes in the custom design and construction of a variety of pooled libraries for commonly used CRISPR applications including knockout libraries for functional screens of coding genes, CRISPRa libraries for gain-of-function screens of coding genes or regulatory function screens of noncoding regions and CRISPRi libraries for loss-of-function screens of coding genes or for screening regulatory function of noncoding regions. Additionally, we can also build CRISPR barcode libraries for single cell-based screens and other pooled libraries utilizing CRISPR technologies.
We can deliver your library as an E. coli stock, plasmid DNA pool, or packaged virus, depending on your needs. Our custom libraries are fully validated by next-generation sequencing so that you know exactly what you get.
Click here for detailed information on our library construction services
In addition to custom pooled CRISPR libraries, we offer premade dual-gRNA lentivirus libraries for whole-genome knockout screens in human and mouse. Dual-gRNA libraries are far more powerful than single-gRNA libraries for knockout screens because the introduction of large deletions by a pair of gRNAs targeting the same gene can have much higher efficiencies than a single gRNA in generating loss-of-function mutations. The human and mouse dual-gRNA libraries are made in the form of ready-to-use high-titer pooled lentivirus targeting 20,048 human and 20,493 mouse genes, respectively. Wherever possible, each gene is targeted redundantly by 4-6 different gRNA pairs in separate vectors.
Price and turnaround of premade dual-gRNA productsView more
| Product name | No. of genes | No. of gRNA pairs | Scale | Catalog No. | Price (USD) |
|---|---|---|---|---|---|
| Human Whole-Genome Dual-gRNA Lentivirus Library | 20,048 | 91,926 | Medium (>1.0x108 TU/ml, 1 ml) |
LVM(Lib190505-1046fgb) |
$2,999
|
| Plus (>1.0x108 TU/ml, 5 ml) |
LV5M(Lib190505-1046fgb) |
$3,499
|
|||
| Mouse Whole-Genome Dual-gRNA Lentivirus Library | 20,493 | 90,344 | Medium (>1.0x108 TU/ml, 1 ml) |
LVM(Lib190505-1050kpm) |
$2,999
|
| Plus (>1.0x108 TU/ml, 5 ml) |
LV5M(Lib190505-1050kpm) |
$3,499
|
Click here for detailed information on our premade dual-gRNA CRISPR libraries
CRISPR solutions
The versatility of the CRISPR system makes it suitable for a wide variety of genome editing applications in mammalian cells. Our technical team with extensive experience in molecular biology applications, can provide complete solutions for all your CRISPR genome editing projects from experimental design to generation of ready-to-use reagents. We can help you with common CRISPR applications listed below, or we can work with you on any novel CRISPR application. Send us a design request today to get a free service proposal!
-
Introduction of point mutation
-
Fragment knockin
- Small tag insertion (< 60 bp)
- Large fragment insertion (up to 4-5 kb)
-
CRISPR gene activation
-
CRISPR gene inhibition
-
CRISPR libraries (KO, CRISPRa, CRISPRi, barcoding, etc.)
-
Stable cell line generation
- Cas9-expressing cell line
- iPSC genome editing
- Cancer cell genome editing
Click here for detailed information on our stable cell line generation services
Technical Information
CRISPR-mediated genome editing
The conventional use of CRISPR system contains two components: Cas9 protein and guide RNA (gRNA). The most commonly used Cas9 is engineered from Streptococcus pyogenes (a.k.a. SpCas9 or hCas9). It is an RNA-guided DNA nuclease which can generate double-stranded breaks (DSBs) at target sites (Figure 1). Another widely used Cas9 variant, Cas9 “nickase” (e.g. Cas9(D10A)), generates single-stranded cuts in DNA. If Cas9 nickase is used in conjunction with two gRNAs targeting the two opposite strands flanking a single target region, the nickase will generate two single-stranded cuts, one on each strand, resulting in DSBs at the target region. Once DSBs are generated by the CRISPR system, cells activate the non-homologous end joining (NHEJ) pathway to repair the DNA breaks, which usually results in small random deletions, or more rarely insertions and base substitutions. When these mutations disrupt a protein-coding region (e.g. a deletion causing a frameshift), they may lead to functional gene knockout.

Figure 1. Mechanisms of CRISPR-induced DNA repair.
Alternatively, and less efficiently, DSBs can be repaired via the homology-directed repair (HDR) pathway, when an exogenous donor DNA template is co-introduced with the CRISPR components. This can result in replacement of the target genomic DNA sequence with donor sequence, generating precise sequence changes such as point mutations or knockin of the donor DNA sequence at the target site. The donor DNA template can be a single-stranded oligo nucleotide (ssODN) or a dsDNA fragment (usually linearized plasmid DNA). ssODNs are suitable for introducing point mutations or small tag insertions, while dsDNA fragments are widely used to introduce large fragment knockins.

Figure 2. Generating homozygous CD274 knockout (KO) mutants using the gRNA-Cas9 ribonucleoprotein (RNP) approach. (A) The editing RNP is electroporated into target cells, and single clones are isolated and screened. The genotypes of the candidates are validated using PCR and Sanger sequencing. (B) In this case study of editing a murine colon adenocarcinoma cell line, cells were electroporated with RNP binding to two sites on the targeted gene to KO a 13-kbp region. Four primers, P1 to P4, were used in three PCR to differentiate KO and WT clones. Based on the (C) PCR results, clone 1 are validated to be homozygous KO mutants, which is also confirmed by (D) sequencing results.
CRISPR-mediated gene regulation
Catalytically inactive form of the Cas9 (a.k.a. dCas9) can be combined with different transcriptional complexes to achieve CRISPR-mediated transcriptional regulation of endogenous genomic loci. To activate gene transcription, the Synergistic Activation Mediator (SAM) system can be used. This system utilizes three components: dCas9/VP64 fusion protein, MS2/P65/HSF1 helper, and a modified gRNA. The dCas9 protein is engineered with VP64, a synthetic transcriptional activator, and the gRNA is modified to include MS2 aptamers which recruit the MS2 complex consisting of the transactivation domains of NF-kB (p65) and HSF1. VP64 consists of four tandem repeats of herpes simplex virus protein 16 and is fused to the C-terminus of the Cas9 protein which results in the recruitment of transcriptional machinery that activates gene expression. The transactivation domains of p65 and HSF1 are potent transcriptional activators that act to further recruit cofactors involved in transcription to promoters or enhancers. Together, the dCas9/VP64 and MS2/P65/HSF1 complexes work synergistically to activate target gene expression, as shown in Figure 2. Beyond the SAM system, VectorBuilder offers the dCas9-VP64-p65-Rta (VPR) transactivation domain system which replaces the transactivation domain of HSF1 with the Epstein-Barr virus R transactivator.
In contrast, to repress target gene transcription, dCas9 can be engineered with a transcriptional repressor, such as a Kruppel-associated box domain (KRAB) and MeCP2. Two versions of the dCas9/KRAB helper vector are currently available: the original dCas9/KRAB helper vector version and an improved dCas9/KRAB/MeCP2 helper vector version which drives the expression of dCas9 fused to a bipartite repressor domain, KRAB/MeCP2, for achieving more potent transcriptional repression of DNA target sites. When gRNA recruits dCas9/KRAB/MeCP2 to an endogenous promoter, as in Figure 3, transcription is repressed. The KRAB domain is commonly found in human transcription factors and is one of the most potent transcriptional repressors currently identified in the human genome. MeCP2 further enhances KRAB-mediated repression through recruitment of histone deacetylases, resulting in chromatin condensation and epigenetic-mediated repression. CRISPR-mediated gene regulation can be used for a wide variety of applications including testing regulatory elements on chromatin, regulating target genes of interest, or for whole genome screening.
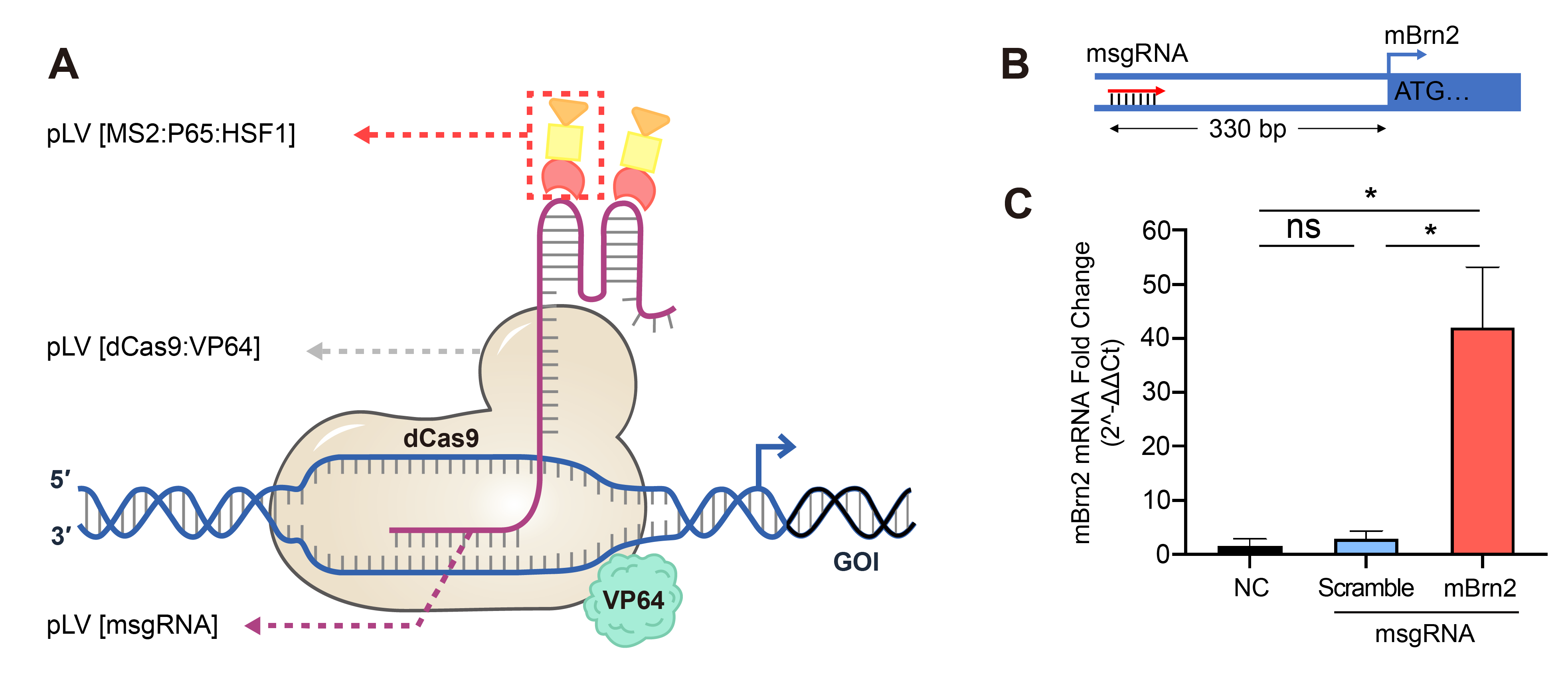
Figure 3. Up-regulation of gene expression achieved by the lentivirus-based CRISPRa. NIH3T3 cells stably expressing SAM complex, dCas9/VP64, and MS2/P65/HSF1 were transduced with msgRNA expression lentivirus followed by antibiotic selection. (A) Illustration of SAM system regulated transcriptional activation. (B) Diagram of msgRNA design targeting the promoter region of the mouse Brn2 gene. (C) Relative gene expression of Brn2 in NIH3T3 cells transduced with scramble or targeting msgRNA or no treatment control (NC), measured by qRT-PCR. Mean±SD, *P<0.05, ANOVA with Tukey’s post hoc test.

Figure 4. Down-regulation of gene expression achieved by the lentivirus-based CRISPRi. Jurkat cells stably expressing the dCas9/KRAB/MeCP2 transcriptional repressor complex were transduced with gRNA expression lentivirus followed by antibiotic selection. (A) Illustration of dCas9/KRAB/MeCP2 regulated gene transcriptional inhibition. (B) Diagram of gRNA design targeting the promoter region of the human CXCR4 gene. (C) CXCR4 protein levels in Jurkat cells transduced with scramble or targeting gRNA or no treatment control (NC), measured by western blot. (D) Relative CXCR4 gene expression in Jurkat cells transduced with scramble or targeting gRNA or no treatment control (NC), measured by qRT-PCR. Mean±SD, ***P<0.001, ****P<0.0001, ANOVA with Tukey’s post hoc test. (E) The surface expressed CXCR4 in the Jurkat cells transduced with scramble or targeting gRNA were quantified by flowcytometry. CXCR4 was labeled with monoclonal primary antibodies (Ab) and fluorophore-conjugated secondary Ab. Unlabeled and secondary Ab only Jurkat cells were used as negative controls. (F) The amount of CXCR4 on the surface of cells transduced with the CXCR4 targeting gRNA was averagely reduced by about 50% compared to the cells transduced with the scramble gRNA. Mean±SD.
CRISPR delivery approaches
CRISPR components can be introduced into mammalian cells via different approaches (Figure 4), including:
- gRNA and Cas9 plasmid
- gRNA and Cas9 virus (i.e. lentivirus, AAV, adenovirus, etc.)
- Mixture of gRNA and Cas9 mRNA
- Preformed RNP between gRNA and Cas9 protein
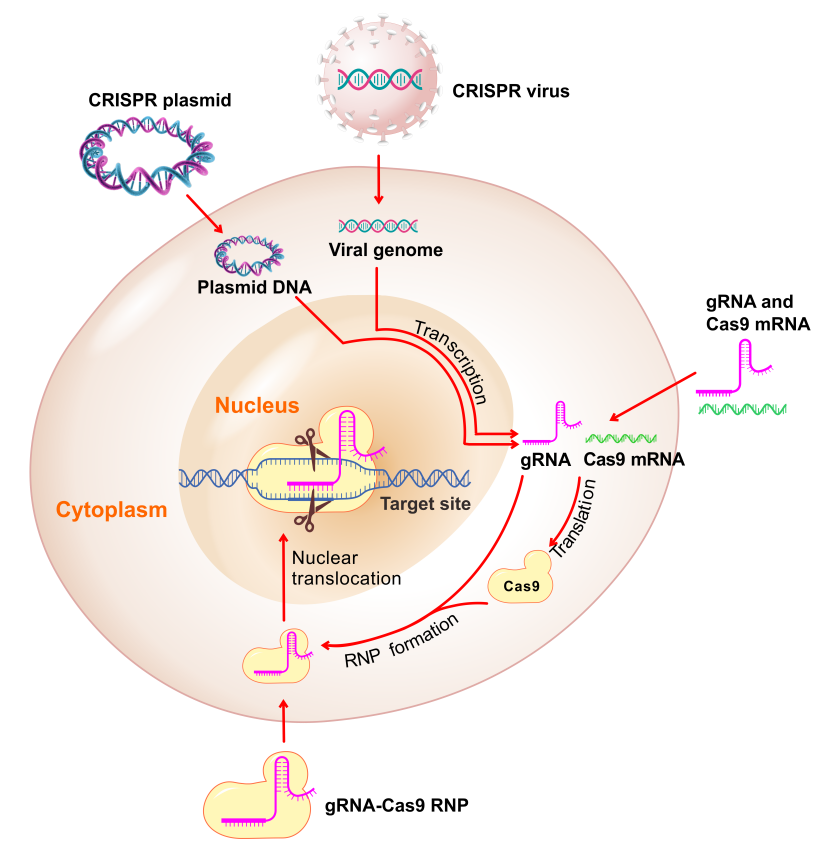
Figure 5. Methods for delivering CRISPR components into cells.
The table below lists key advantages and disadvantages for each delivery approach and can be used as a reference to help you decide the most suitable approach for your experiments:
| Delivery Approach | Advantages | Disadvantages |
|---|---|---|
| gRNA and Cas9 plasmid |
|
|
| gRNA and Cas9 virus |
|
|
| Mixture of gRNA and Cas9 mRNA |
|
|
| Preformed gRNA-Cas9 RNP complex |
|
|
gRNA databases
VectorBuilder’s online CRISPR vector design tool features optimized, whole-genome gRNA databases for human, mouse and rat enabling you to design CRISPR vectors with high targeting efficiency. We follow the algorithm utilized in CRISPR library design (CLD) to calculate specificity scores for gRNAs. Briefly, for a given gRNA intended to target a N(20)NGG sequence in a species, we search for all potential off-target sites in the genome of that species that have ≤3 mismatches with the target sequence. For each potential off-target site identified this way, a single off-target score is calculated. Scores for all the off-target sites are then used in aggregate to calculate the final specificity score of the gRNA, which is between 0 and 100, with higher values indicating greater targeting specificity. Please note that specificity scores are only a rough guide. Actual targeting efficiency and specificity could depart from what the scores predict. gRNAs with low scores may still work well.
When you design CRISPR vectors on VectorBuilder’s online platform, you will have the option to search for your target genes in our database. Upon entering your gene name, you will see detailed information on all available guide RNA designs against your target gene available in our database. Our whole-genome gRNA database allows you to easily pick appropriate guide sequence for your target genes without looking for and analyzing the target sites on your own, thereby providing you with advantages offered by popular gRNA design tools or software.
VectorBuilder's online “Learning Center” under "Resources" contains rich educational resources to help you to successfully plan, execute and troubleshoot your CRISPR experiments.
Click here to read guides on CRISPR vector systemsClick here to read guides on CRISPR components
Resources
FAQ
Either shRNA-mediated knockdown or nuclease-mediated knockout (e.g. CRISPR or TALEN) can be valuable experimental approach to study the loss-of-function effects of a gene of interest in cell culture. In order to decide which method is optimal for your specific application, there are a few things you should consider.
Mechanisms
Knockdown vectors
Knockdown vectors express short hairpin RNAs (shRNAs) that repress the function of target mRNAs within the cell by inducing their cleavage and repressing their translation. Therefore, shRNA knockdown vectors are not associated with any DNA level sequence change of the gene of interest.
Read more about our shRNA knockdown vectors
Knockout vectors
CRISPR and TALEN both function by directing nucleases to cut specific target sites in the genome. These cuts are then inefficiently repaired by the cellular machinery, resulting in permanent mutations, such as small insertions or deletions, at the sites of repair. A subset of these mutations will result in loss of function of the gene of interest due to frame-shifts, premature stop codons, etc. If two closely positioned cut sites in the genome (i.e. within several kb) are targeted simultaneously, this can also result in the deletion of the intervening region.
Read more about our CRISPR vectors
Effectiveness
shRNA-mediated knockdown will never completely repress the expression of the target gene. Even for the most effective shRNAs, some residual expression of the target gene will remain. In contrast, in a fraction of treated cells, CRISPR and TALEN can generate permanent mutations which may result in complete loss of gene function.
Consistency and uniformity
shRNA vectors generally provide high cell-to-cell uniformity within the pool of treated cells and very consistent results between experiments. In contrast, CRISPR and TALEN produce results that are highly non-uniform from cell to cell due to the stochastic nature of the mutations introduced. To fully knock out the gene of interest in a cell, all copies of the gene in the cell must be knocked out. Given that normal cells have two copies of any gene (except for X- or Y-linked genes) while cancer cells can have more than two copies, such full knockout cells may represent a very small fraction of all the treated cells. For this reason, nuclease-mediated knockout experiments require the screening of clones by sequencing to identify the subset in which all copies of the gene of interest have been knocked out.
Off-target effects
Off-target effects have been reported for both shRNA-mediated knockdown and nuclease-mediated knockout. The off-target phenotype(s) can be estimated by using multiple different shRNAs to target the same gene. If a gene knocked down by multiple different shRNAs results in consistent phenotype(s), then it argues against the phenotype(s) being caused by off-target effects. For CRISPR- or TALEN-mediated knockout, multiple clones containing loss-of-function mutations should be analyzed in order to account for any phenotype(s) that may be due to off-target mutations. Additionally, bioinformatically identified off-target sites could be sequenced in the clones to see if they have been mutated.
Both CRISPR and TALEN systems have been harnessed to edit genomes of cultured cells and model organisms. Both systems can be used to knock out genes, or to knock in point mutations or insertions, but these two systems are different in several ways and have their own pros and cons.
Mechanisms
CRISPR
The CRISPR system uses a site-specific guide RNA (gRNA) to direct the Cas9 nuclease to its target site in the genome to create DNA cleavage. The target sequence is typically ~20 bp long, and sites containing a few mismatches may still be recognized and cleaved.
Read more about our CRISPR vectors
TALEN
The TALEN system employs a pair of chimeric proteins, each composed of a TAL effector DNA-binding domain (recognizing a specific sequence) fused to a FokI nuclease domain. The pair of proteins are designed to bind to a pair of target sites in the genome, each ~18 bp long and flanking a 14-20 bp spacer. Upon binding to DNA, the Fokl nuclease domains on the pair of proteins are able to dimerize, which in turn leads to DNA cleavage within the spacer region between the two target sites.
Efficiency
Both CRISPR- and TALEN-mediated genome editing show good efficiency, but the efficiency varies a lot depending on application, species and cell type. In general, CRISPR can be delivered into cells and induce DNA cleavage more efficiently than TALEN.
Off-target effects
A CRISPR gRNA targets ~20 bp sequence, whereas a TALEN pair binds to a total of ~36 bp target sequence. In addition, Cas9/gRNA complex has higher tolerance for sequence mismatches (up to 5 bp mismatches) than TALEN does. Therefore, TALEN-mediated cleavage has better specificity than CRISPR, and off-target cleavage in the genome by TALEN is unlikely. In contrast, off-target effects have been reported for CRISPR in cell lines, though analyses of CRISPR knockout mice suggest lower off-target frequency in vivo. Recent developments of CRISPR system have significantly enhanced CRISPR specificity. By using Cas9 nickase (Cas9 mutant that contains only one catalytic nuclease domain, e.g. Cas9_D10A and Cas9_H840A) with dual gRNAs, two single-strand DNA nicks are generated with close proximity of the target region, resulting in a double-nick DSB (double-strand break) within the target region that could be repaired. In this design, the off-target effects are minimized since the dual gRNAs expand the target sequence to ~40 bp long.
Target site requirements
TALEN can be generated to specifically target nearly any sequence in the genome. In contrast, target site selection for CRISPR is limited by the requirement for a PAM sequence (typically NGG) sequence located on the immediate 3’ end of the gRNA target sequence. This is no barrier to knocking out genes because cleavage anywhere in the gene is potentially effective but may present difficulties in generating site-specific mutations or insertions that require cleavage at a specific position of the gene. To precisely edit a specific genomic site using CRISPR, a homologous recombination donor vector or long oligo containing the desired edit sequence flanking by the immediate upstream and downstream homology arms of the target site can be delivered to the cells together with gRNA(s) and Cas9, in order to guide HDR (homology directed repair)-mediated DNA repair at the target site.
Design your homologous recombination donor vector online
Technical challenges
In terms of technical simplicity, CRISPR out competes TALEN in several ways. First, for vector construction, CRISPR system only needs to construct a short gRNA because targeting of Cas9/gRNA complex relies on simple RNA/DNA hybridization, while TALEN system requires re-engineering of the TAL DNA-binding domain that is unique for each protein-DNA interaction. Therefore, gRNAs are cheaper and easier to design and construct than TALENs which always require two vectors per target site. Secondly, for some applications, such as injecting mouse embryos, Cas9 protein and gRNA can be more efficiently delivered via direct injection, but TALEN cannot. Thirdly, CRISPR is extremely versatile in genetic screening experiments since CRISPR screening library expressing many thousands different gRNAs can be easily constructed in a high-throughput manner.
For CRISPR-mediated genome editing, Cas9 nuclease is directed to the target site of site-specific guide RNA (gRNA) in the genome to create DNA cleavage. In most cases, to generate simple gene knockout, a single gRNA can be used together with Cas9 to generate a double-strand break (DSB), which is then inefficiently repaired by the non-homologous end joining (NHEJ), resulting in permanent mutations, such as small insertions or deletions, at the site of repair. A subset of these mutations will result in loss of function of the gene of interest due to frame-shifts, premature stop codons, etc.
Dual gRNAs can be used if Cas9_D10A nickase is being used to target the two opposite strands of a single target site. In this approach, the nickase enzyme will generate single strand cuts on both strands, one guided by each of the two gRNAs, resulting in DSBs at the target site. Generally, this method reduces off-target effects of CRISPR/Cas9 expression because targeting by both gRNAs is necessary for DSBs to be generated.
Dual gRNAs can also be used when Cas9_D10A nickase and an exogenous donor DNA template are being used to introduce specific base-changes (e.g. knockins) into a gene of interest. In this approach, the two opposite strands would be targeted by the two gRNAs at two sites flanking the desired mutation site, and homology-directed repair (HDR) pathways make use of the exogenous donor template to repair the excised sequence.