



No SMAll Feat
Keywords: Zolgensma mechanism, spinal muscular atrophy
Spinal muscular atrophy (SMA) is a rare genetic neuromuscular disorder characterized by the loss of motor neurons in the spinal cord and brainstem, leading to progressive muscle weakness and atrophy. SMA is caused by mutations in the survival motor neuron 1 (SMN1) gene, which is responsible for producing the SMN protein essential for motor neuron function. Recently, two genetic therapies, Spinraza and Zolgensma, have emerged as prominent treatments for the disease and work by two unique mechanisms; in this article we will review SMA from a genetic perspective and then dive into the mechanisms behind these therapies.
Prone to error
The SMN1 gene encodes a protein that is involved in various RNA processes including splicing and metabolism. A near identical gene, SMN2, is situated ~1000 kbs closer to the centromere of the chromosome. However, small differences in the coding sequence lead to significant functional differences. One key distinction between the two genes is a nucleotide at a splice junction of exon 7, with SMN1 retaining the nucleotide required for efficient exon 7 inclusion, while SMN2 harbors a nucleotide substitution that leads to preferential skipping of exon 7 during mRNA processing. This difference results in SMN1 producing a full-length, functional protein, while SMN2 predominantly generates a truncated and unstable protein.
The SMN1 gene is in a region of chromosome 5 that contains highly repetitive sequences and is prone to genomic rearrangements, such as deletions or duplications. Additionally, the high instability of this genomic region can result in different types of mutations, including large-scale deletions of the SMN1 gene or duplications of nearby genes. The presence of repetitive sequences, such as retrotransposons and other repetitive elements, makes the SMN1 gene locus susceptible to non-allelic homologous recombination (NAHR) events. NAHR can occur during meiosis when homologous regions misalign, leading to unequal crossing over between repetitive sequences. This process can result in the loss or duplication of the SMN1 gene, ultimately affecting the production and function of the SMN protein.
The repetitive nature of the region increases the likelihood of NAHR events between SMN1 and SMN2, leading to misalignment and subsequent deletion of exon 7. This is the most common mutation in SMA.
Spinraza
Spinraza, the first approved therapeutic for the treatment of spinal muscular atrophy (SMA), is an antisense oligonucleotide (ASO) therapy that received FDA approval in 2016, marking a significant milestone in addressing this disease. The ASO is designed to bind to a specific region of the SMN2 pre-messenger RNA which alters the splicing process leading to inclusion of exon 7 during RNA processing. As a result, more mature mRNA molecules are produced with intact exon 7, allowing for the synthesis of a full-length, functional SMN protein which helps compensate for the deficiency caused by mutations or deletions in the SMN1 gene.
Spinraza is administered directly into the patient's cerebrospinal fluid through a lumbar puncture procedure. The treatment protocol consists of an induction phase, during which patients receive more frequent doses, followed by a maintenance phase with fewer treatments. Notably, Phase 3 clinical trials of Spinraza have shown significant improvements in motor function for both infants with SMA and individuals with later-onset SMA when compared to a placebo. As a result, Spinraza has gained widespread acceptance and has been adopted as a standard therapy for SMA treatment worldwide.
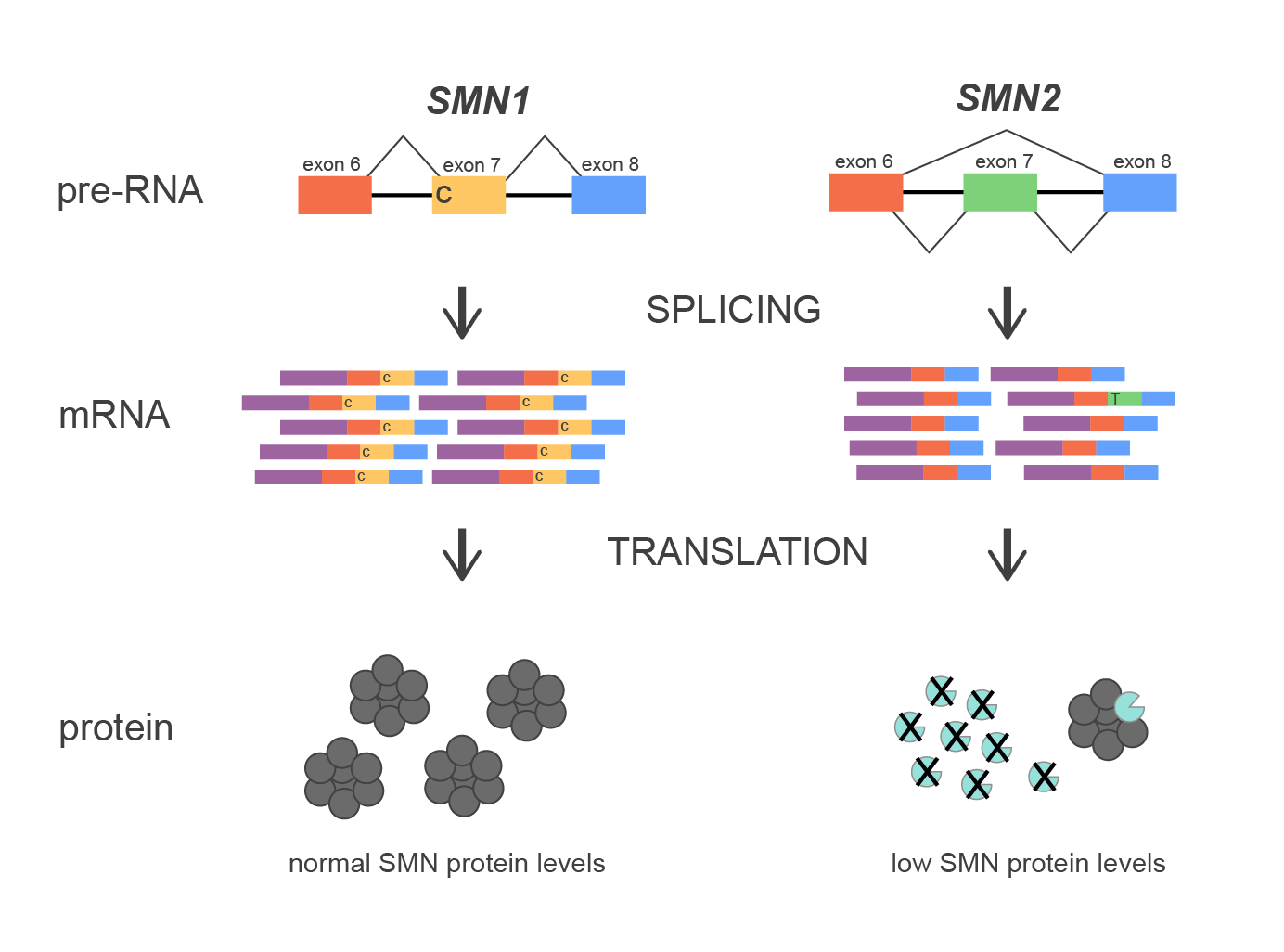
Figure 1. Diagram of splicing mechanism of SMN1 and SMN2 genes
Zolgensma
Zolgensma is a groundbreaking gene therapy that harnesses the power of adeno-associated virus (AAV) vectors to treat SMA. The mechanism of action of Zolgensma is to deliver a functional copy of the SMN1 gene and thus address the underlying cause of SMA.
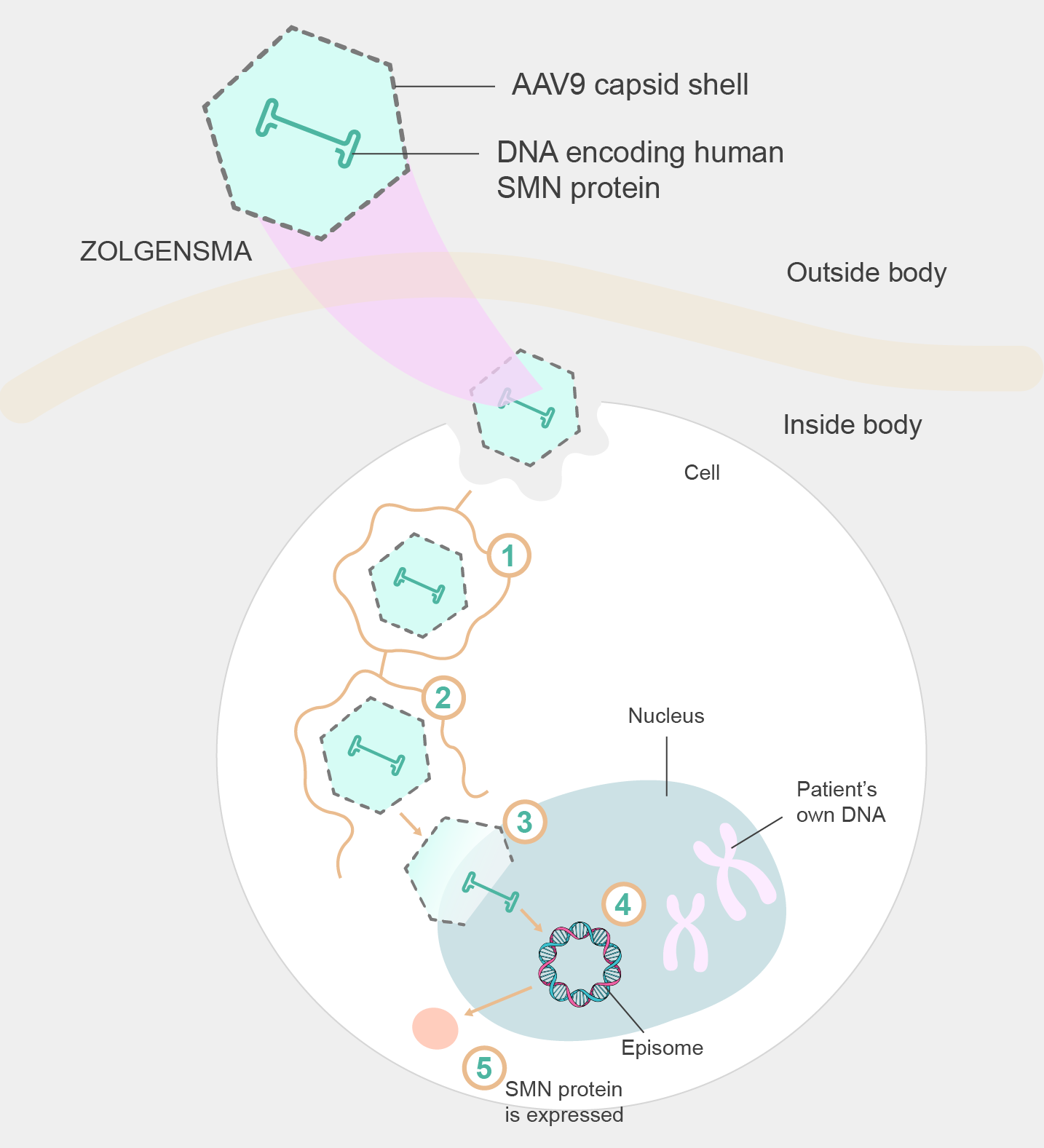
Figure 2. Mechanism of Zolgensma delivery
AAV vectors have become a leading choice in gene therapy due to their natural ability to efficiently infect and transfer genetic material into target cells. Different AAV serotypes can be utilized to target specific tissues/cells. For example, AAV9 exhibits a preference for crossing the blood-brain barrier and efficiently targeting motor neurons, making it an ideal candidate for delivering the SMN1 gene to the affected cells in SMA patients. The AAV9 capsid, the protein shell that encapsulates the viral genome, protects the therapeutic cargo during delivery and aids in cellular entry and transduction.
By utilizing AAV9 as the vector for Zolgensma, the therapy aims to restore SMN protein levels in motor neurons, thereby preventing further degeneration and improving motor function in patients with SMA. The AAV9 vector delivers the functional SMN1 gene directly into the target cells, where it initiates the production of the SMN protein. This mechanism allows Zolgensma to address the root cause of SMA at the genetic level, potentially offering long-term therapeutic benefits. The choice of AAV9 as the vector for Zolgensma reflects a strategic selection based on its ability to target motor neurons efficiently and specifically, making it a critical component of the therapy's success in treating SMA.
Zolgensma is administered to patients through a one-time intravenous infusion. The exact dose is determined by a person’s body weight and the administration process can take several hours, during which the patient is monitored for any adverse reactions. Due to the nature of AAV vectors, expression of SMN1 in neurons may not be detected for a few days to a couple of weeks. Phase 1, 2, and 3 clinical trials for Zolgensma demonstrated consistent and encouraging results. In Phase 1, a small trial involving infants with SMA Type 1, Zolgensma showed a rapid increase in survival rates and motor milestones, with some infants achieving the ability to sit independently. Phase 2 trials further validated these findings, revealing sustained motor function improvements and a delay in disease progression. In Phase 3, Zolgensma-treated infants displayed dramatic motor skill improvements, and a high percentage achieved the ability to sit without support.
Comparing the options
Zolgensma is delivered through a one-time intravenous infusion, introducing a functional copy of the SMN1 gene into motor neurons to address the genetic cause of SMA. In contrast, Spinraza is an antisense oligonucleotide administered via regular spinal injections, modifying the splicing of the SMN2 gene to increase the production of the SMN protein and compensate for the lack of functional SMN1 gene. Zolgensma and Spinraza significantly differ in cost. Zolgensma, being a one-time gene therapy, comes with a higher upfront cost, while Spinraza, administered regularly, accumulates costs over time but is relatively cheaper in terms of individual treatment expenses.
Spinraza and Zolgensma have distinct side effect profiles and toxicity considerations. Spinraza, as an antisense oligonucleotide, may cause side effects related to repeated spinal injections, such as injection site pain, discomfort, and potential risks of complications associated with the procedure. On the other hand, Zolgensma, being a gene therapy, has demonstrated a generally favorable safety profile in clinical trials; however, it may carry the risk of immune responses to the viral vector used for gene delivery, which requires careful monitoring and management.
When deciding between Spinraza and Zolgensma for a patient with SMA, a clinician would consider several factors. Firstly, they would evaluate the patient's age, disease severity, and overall health status, as both treatments may have varying efficacy in different SMA types and stages. For infants and young children with severe SMA, Zolgensma's one-time gene therapy could be a compelling option due to its potential for rapid motor function improvements and the possibility of achieving developmental milestones. However, for older or less severe SMA patients, Spinraza's well-established efficacy and safety profile may be more suitable. Additionally, the accessibility of the treatments, insurance coverage, and the patient's individual preferences would also be considered during the decision-making process. Ultimately, the clinician's expertise and a thorough understanding of the patient's unique circumstances would play a crucial role in making the most appropriate and personalized treatment recommendation.
In conclusion, spinal muscular atrophy (SMA) is a complex genetic neuromuscular disorder characterized by the loss of motor neurons and caused by mutations in the SMN1 gene. The development of two prominent treatments, Spinraza and Zolgensma, has brought renewed hope for patients with SMA. Both Spinraza and Zolgensma represent significant advancements in treating SMA and provide new avenues for managing this challenging disease. Further research and ongoing clinical studies will continue to refine and optimize these therapies, potentially improving outcomes and quality of life for patients with SMA.
Sources
Fischell JM, Fishman PS. A Multifaceted Approach to Optimizing AAV Delivery to the Brain for the Treatment of Neurodegenerative Diseases. Front Neurosci. 2021 Sep 24;15:747726. doi: 10.3389/fnins.2021.747726. PMID: 34630029; PMCID: PMC8497810.
https://www.spinraza.com/en_us/home/why-spinraza/how-spinraza-works.html?cid=PPC-GOOGLE-Branded_Zolgensma_Exact_Tier+1~S~PH~BR~NER~DTC~COM-zolgensma-NA-p74193523522&gclid=EAIaIQobChMIhsCT3YaKgAMVmOqUCR1C2w3bEAAYASAAEgL8cvD_BwE&gclsrc=aw.ds